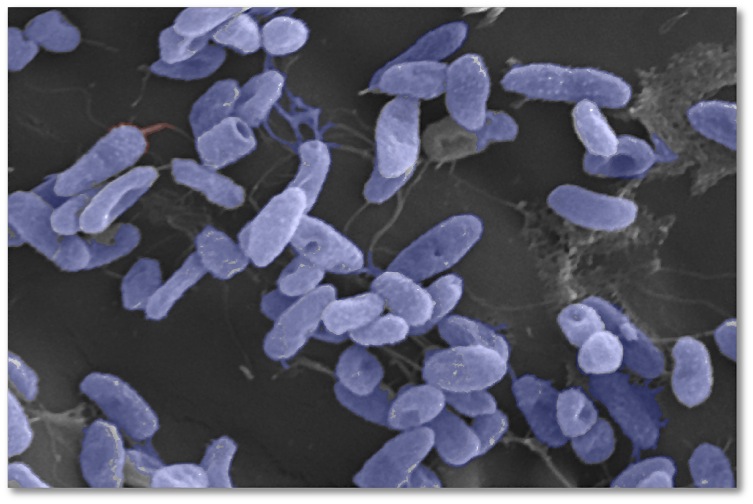
 At the Laboratory for Microbial and Environmental Genomics, we investigate how microorganisms respond to changes in their ecosystem. Two primary areas of research are 1) assessing how microbes respond to plastic pollution and 2) assessing how microbes respond to sewage and nutrient pollution. These pervasive contaminants have a detrimental impact on coastal ecosystems, leading to habitat loss and the disruption of marine food webs. Research outcomes include water quality remediation and habitat restoration. We also explore links between the ocean and human health by investigating the ecology and virulence of marine microbes that are opportunistic human pathogens. Click on the publications tab to learn more about our research, and follow us on Twitter to receive updates about these projects.
At the Laboratory for Microbial and Environmental Genomics, we investigate how microorganisms respond to changes in their ecosystem. Two primary areas of research are 1) assessing how microbes respond to plastic pollution and 2) assessing how microbes respond to sewage and nutrient pollution. These pervasive contaminants have a detrimental impact on coastal ecosystems, leading to habitat loss and the disruption of marine food webs. Research outcomes include water quality remediation and habitat restoration. We also explore links between the ocean and human health by investigating the ecology and virulence of marine microbes that are opportunistic human pathogens. Click on the publications tab to learn more about our research, and follow us on Twitter to receive updates about these projects.